

- Sie haben noch keine Artikel in Ihrem Warenkorb.
SARS-CoV-2 infection and persistence in the human body and brain at autopsy (News)
Abstract
Coronavirus disease 2019 (COVID-19) is known to cause multi-organ dysfunction1,2,3 during acute infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), with some patients experiencing prolonged symptoms, termed post-acute sequelae of SARS-CoV-2 (refs. 4,5). However, the burden of infection outside the respiratory tract and time to viral clearance are not well characterized, particularly in the brain3,6,7,8,9,10,11,12,13,14. Here we carried out complete autopsies on 44 patients who died with COVID-19, with extensive sampling of the central nervous system in 11 of these patients, to map and quantify the distribution, replication and cell-type specificity of SARS-CoV-2 across the human body, including the brain, from acute infection to more than seven months following symptom onset. We show that SARS-CoV-2 is widely distributed, predominantly among patients who died with severe COVID-19, and that virus replication is present in multiple respiratory and non-respiratory tissues, including the brain, early in infection. Further, we detected persistent SARS-CoV-2 RNA in multiple anatomic sites, including throughout the brain, as late as 230 days following symptom onset in one case. Despite extensive distribution of SARS-CoV-2 RNA throughout the body, we observed little evidence of inflammation or direct viral cytopathology outside the respiratory tract. Our data indicate that in some patients SARS-CoV-2 can cause systemic infection and persist in the body for months.
Main
COVID-19 has respiratory and non-respiratory manifestations1,2,3, including multi-organ failure and shock among patients with severe and fatal disease. Some individuals who survive experience post-acute sequelae of SARS-CoV-2, also known as long COVID4,5. Although autopsy studies of fatal COVID-19 cases support the ability of SARS-CoV-2 to infect multiple organs3,7,8,9,10,11,12, extrapulmonary organs often lack histopathological evidence of virally mediated injury or inflammation10,11,12,13,14. The paradox of infection outside the respiratory tract without injury or inflammation raises many pathogen- and host-related questions.
To investigate the cellular tropism, replication competence, persistence and evolution of SARS-CoV-2 in humans, and to look for associated histopathology in infected tissues, we carried out autopsies on 44 COVID-19 cases. Our approach focused on timely, systematic and comprehensive tissue sampling and preservation for complementary analyses. We carried out droplet digital polymerase chain reaction (ddPCR) for detection and quantification of SARS-CoV-2 nucleocapsid (N) gene targets and in situ hybridization (ISH) to validate the ddPCR findings and determine the cellular tropism of SARS-CoV-2. Immunofluorescence (IF) and chromogenic immunohistochemistry (IHC) were used to further validate the presence of SARS-CoV-2 in the brain. We carried out quantitative real-time PCR with reverse transcription (RT–qPCR) to detect subgenomic RNA, a marker suggestive of recent virus replication15, and demonstrated replication-competent SARS-CoV-2 in selected respiratory and non-respiratory tissues, including the brain, by virus isolation in traditional and modified Vero E6 cell culture. In six individuals, we measured the diversity and anatomic distribution of intra-individual SARS-CoV-2 spike gene variants using high-throughput, single-genome amplification and sequencing (HT-SGS).
We categorized autopsy cases as early (n = 17), mid (n = 13) or late (n = 14) by illness day (d) at the time of death, being ≤d14, d15–30 or ≥d31, respectively. We defined persistence as the presence of SARS-CoV-2 RNA among late cases. We analysed and described our results in terms of respiratory and non-respiratory tissues to quantify and statistically compare SARS-CoV-2 RNA levels across tissues and cases.
Autopsy cohort overview
Between 26 April 2020 and 2 March 2021, we carried out 44 autopsies, all among unvaccinated individuals who had died with COVID-19. SARS-CoV-2 PCR positivity was confirmed premortem in 42 cases and postmortem in 2 cases (P3 and P17; Extended Data Fig. 1). A total of 38 cases were SARS-CoV-2 seropositive (Supplementary Data 1a), 3 were seronegative (P27, P36 and P37), and plasma was unavailable for 3 cases (P3, P4 and P15). Brain sampling was accomplished in 11 cases (Fig. 1). The cohort was racially and ethnically diverse. Thirty per cent were female, and the median age was 62.5 years (interquartile range (IQR): 47.3–71.0; Extended Data Table 1a). A total of 61.4% had three or more comorbidities. The median interval from symptom onset to final hospitalization and subsequently death was 6 days (IQR: 3–10) and 18.5 days (IQR: 11.25–37.5), respectively (Extended Data Table 1b). The median postmortem interval was 22.2 h (IQR: 18.2–33.9). Individual-level case data can be found in Supplementary Data 2a.
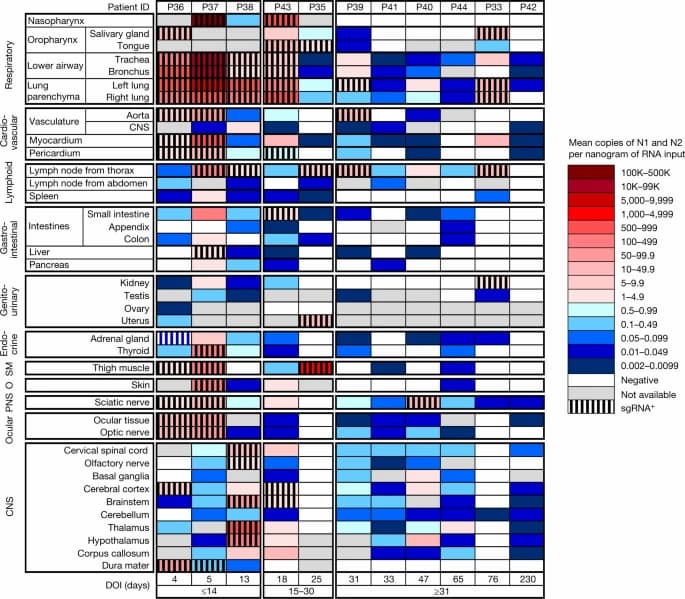
Fig. 1: Distribution, quantification and replication of SARS-CoV-2 across the human body and brain.
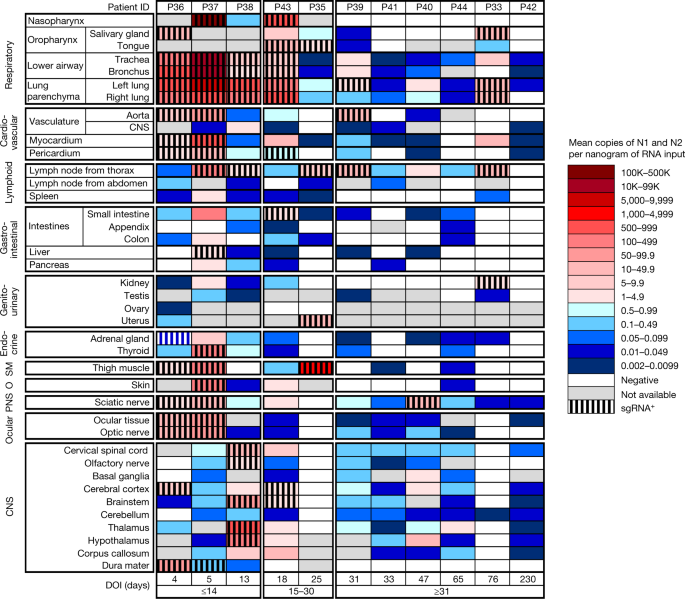
The heat map depicts the highest mean quantification of SARS-CoV-2 RNA (N) through ddPCR present in the autopsy tissues of 11 patients who died with COVID-19 and underwent whole-body and brain sampling. Patients are aligned from shortest to longest duration of illness (DOI) before death, listed at the bottom of the figure, and grouped into early (≤14 days), mid (15–30 days) and late (≥31 days) duration of illness. Tissues are organized by tissue group beginning with the respiratory tissues at the top and CNS at the bottom. Viral RNA levels range from 0.002 to 500,000 N gene copies per nanogram of RNA input, depicted as a gradient from dark blue at the lowest level to dark red at the highest level. Tissues that were also positive for subgenomic RNA (sgRNA+) through real-time RT–qPCR are shaded with black vertical bars. O, other; PNS, peripheral nervous system; SM, skeletal muscle.
Widespread infection and persistence
SARS-CoV-2 RNA was detected in 84 distinct anatomical locations and body fluids (Supplementary Data 1b–d), with a significantly (P < 0.0001 for all) higher burden detected in respiratory compared with non-respiratory tissues among early (2.04 ± 0.10 log10[N gene copies] per nanogram of RNA), mid (1.36 ± 0.12 log10[N gene copies] per nanogram of RNA) and late (0.67 ± 0.11 log10[N gene copies] per nanogram of RNA; Extended Data Fig. 2a) cases. We compared linear trends in SARS-CoV-2 RNA levels by illness day, as a continuous variable, and observed a significantly steeper negative slope of SARS-CoV-2 RNA levels in respiratory (−3.14, s.e. 0.39) compared with non-respiratory (−1.62, s.e. 0.38; P < 0.0001) tissues (Extended Data Fig. 2b,c).
We detected SARS-CoV-2 RNA in perimortem plasma of 11 early and 1 mid case (Supplementary Data 1b,d). SARS-CoV-2 RNA was undetectable or just above the limit of detection in peripheral blood mononuclear cells from select early and mid cases (Supplementary Data 1a). The median and IQR of SARS-CoV-2 N gene copies per nanogram of RNA and proportion of cases with RNA detected in each tissue group and fluids are summarized in Extended Data Table 2. SARS-CoV-2 RNA persistence was detected across multiple tissue groups among all late cases despite being undetectable in plasma in any (Supplementary Data 1b–d). SARS-CoV-2 RNA was detected in central nervous system (CNS) tissue in 10/11 cases (90.9%), including across most brain regions evaluated in 5/6 late cases, including P42 who died at D230 (Fig. 1).
We detected SARS-CoV-2 subgenomic RNA across all tissue groups and multiple fluid types, including plasma, pleural fluid and vitreous humour (Supplementary Data 1a–c). ddPCR and subgenomic RNA RT-qPCR results closely correlated among 1,025 jointly tested samples (ρ = 0.76; 95% confidence interval (CI): 0.73–0.78), particularly among respiratory samples (n = 369, ρ = 0.86; 95% CI: 0.84–0.89), early cases (n = 496, ρ = 0.88; 95% CI: 0.85–0.89) and samples that tested positive by both assays (n = 302, ρ = 0.91; 95% CI: 0.88–0.93; Extended Data Fig. 2d,e). With sensitivity and specificity weighted equally, a ddPCR value of ≥1.47 N copies per nanogram of RNA predicted a positive subgenomic RNA result with 93.0% sensitivity and 91.6% specificity, with a receiver operating characteristic (ROC) area under the curve (AUC) of 0.965 (95% CI: 0.953–0.977; Extended Data Fig. 2f).
We isolated SARS-CoV-2 in Vero E6 cell culture from diverse tissues in and outside the respiratory tract including heart, lymph node, gastrointestinal tract, adrenal gland and eye from early cases (Extended Data Fig. 3a). In total, we isolated virus from 25/55 (45%) specimens tested across four SARS-CoV-2 subgenomic RNA quantification cycle (Cq) value intervals, with decreasing yield with rising Cq interval. Among the 55 samples tested for virus isolation, with sensitivity and specificity weighted equally, a ddPCR value of ≥758 N copies per nanogram of RNA predicted replication-competent virus with 76% sensitivity and 90% specificity (ROC AUC = 0.887; 95% CI: 0.795–0.978), and a subgenomic RNA value of ≥25,069 copies per microlitre of RNA (about Cq 22.40) predicted replication-competent virus with 72% sensitivity and 100% specificity (ROC AUC = 0.915; 95% CI: 0.843–0.987; Extended Data Fig. 3b,c).
We reattempted virus isolation from thalamus and hypothalamus of P38 on Vero E6-TMPRSS2-T2A-ACE2 cells16 and observed a cytopathic effect 48 h after inoculation with thalamus tissue homogenate from P38. RT–qPCR for SARS-CoV-2 envelope (E) genomic RNA was carried out on the tissue homogenate and the supernatant of the virus isolation process at the time of the cytopathic effect, and yielded Ct values of 27.33 and 13.24, respectively.
Viral genome sequencing
We used HT-SGS to analyse SARS-CoV-2 spike gene variant sequences from a total of 46 tissues in 6 individuals (Extended Data Fig. 4). In P27 (D1), P19 (D7) and P18 (D9), no nonsynonymous virus genetic diversity was detected in respiratory and non-respiratory sites despite a high depth of single-molecule sampling. In P27, two virus haplotypes, each with a single synonymous substitution, were preferentially detected in non-respiratory sites including the right and left ventricles and mediastinal lymph node. In P38 (D13), a D80F residue was identified in 31/31 pulmonary but 0/490 brain sequences, and a G1219V residue was restricted to brain variants. SARS-CoV-2 virus isolated from thalamus of P38 through Vero E6-TEMPRSS2-TA2-ACE2 cell culture and subjected to short-read, whole-genome sequencing matched the minor haplotype detected from P38 RNAlater-preserved thalamus and the major haplotype of P38 RNAlater-preserved hypothalamus. A nonsynonymous substitution was also detected in P36 (D4) dura mater, albeit at very low sampling depth (n = 2 sequences), compared with non-CNS tissue.
ISH reveals the cellular tropism of SARS-CoV-2
We validated our ddPCR results by ISH for SARS-CoV-2 spike RNA in respiratory and non-respiratory tissues in selected early, mid and late cases across >35 cell types and hyaline membranes (Figs. 2 and 3, Extended Data Table 3 and Supplementary Data 3). Detailed annotation of SARS-CoV-2 spike RNA ISH-positive cells by tissue, including across multiple brain regions, is provided in Figs. 2 and 3 and Supplementary Data 3.
Fig. 2: RNA in situ (RNAscope) detection of SARS-CoV-2 in extrapulmonary tissues.
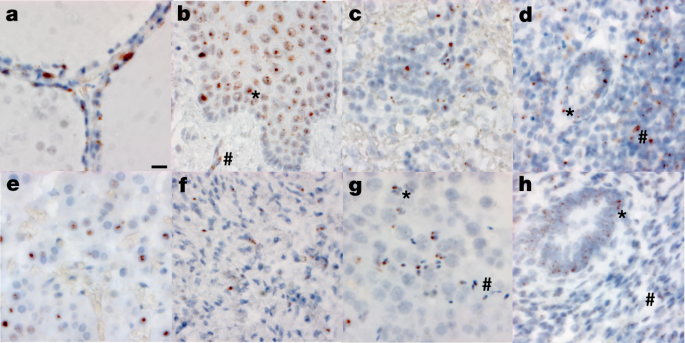
a–h, SARS-CoV-2 virus is localized to the Golgi and endoplasmic reticulum, perinuclear in appearance, in the following organs and cell types (×500 magnifications, scale bars, 2 μm, all panels): thyroid of P19, demonstrating the presence of virus in follicular cells (a), oesophagus of P18, demonstrating the presence of virus in the stratified squamous epithelium (asterisk), as well as signal in capillaries within the stroma (hash) (b), spleen of P19, demonstrating the presence of virus in mononuclear leukocytes in the white pulp (c), appendix of P19, demonstrating the presence of virus in both colonic epithelium (asterisk) and mononuclear leukocytes in the stroma (hash) (d), adrenal gland of P19, demonstrating the presence of virus in endocrine secretory cells (e), ovary of P18, demonstrating the presence of virus in stromal cells of the ovary in a post-menopausal ovary (f), testis of P20, demonstrating the presence of virus in both Sertoli cells (asterisk) and maturing germ cells in the seminiferous tubules of the testis (hash) (g), endometrium of P35, demonstrating the presence of virus in endometrial gland epithelium (asterisk) and stromal cells (hash), in a pre-menopausal endometrial sample (h). The images are exemplars of extrapulmonary tissues that were positive for SARS-CoV-2 N RNA during 20 batches of ISH staining.
S-CoV-2 protein and RNA expression in human CNS tissues.
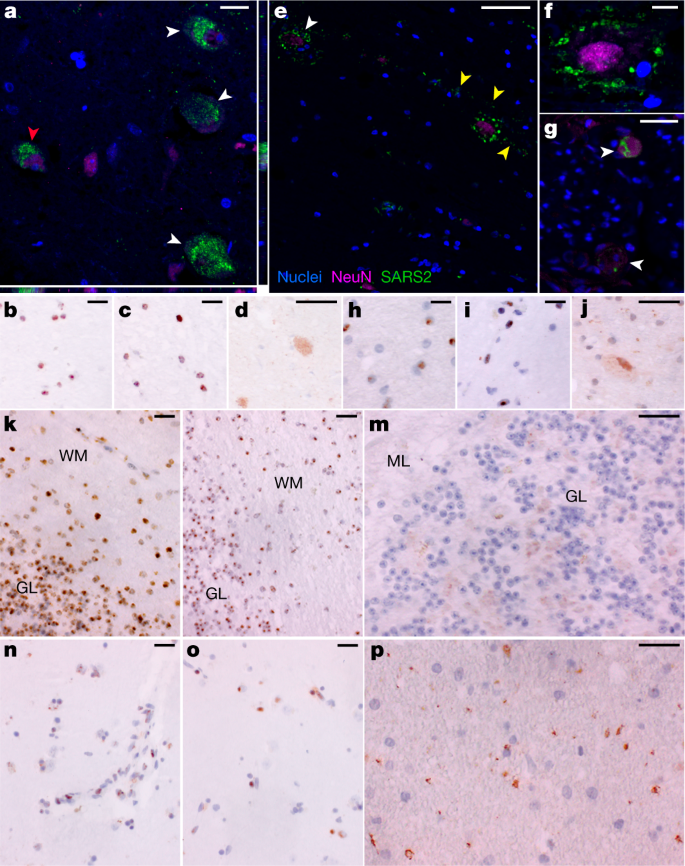
a, High-magnification visualization of hypothalamus from P38 labelled for SARS-CoV-2 N protein (green) and neuronal nuclear (NeuN) protein (magenta), demonstrating viral-specific protein expression in neurons (white arrowheads) by IF. The z-stack orthogonal views to the right and bottom of a demonstrate NeuN labelling in the nucleus and SARS-CoV-2 protein in the cytoplasm of the cell (red arrowhead). b–d, SARS-CoV-2 spike (S) (b) and N (c) RNA (brown) by ISH and SARS-CoV-2 N (d) protein (brown) by chromogenic IHC of hypothalamus of P38. e, Infected neurons were found in the cervical spinal cord of P42 by IF, for which white arrowheads indicate NeuN-positive neurons with associated virus protein. Viral protein labelling was also identified in linear structures radiating away from neuronal cell bodies suggestive of neuronal projections (yellow arrowheads). f, A higher magnification of neuron-associated viral protein labelling. g, Viral protein was also detected by IF in neurons of the spinal ganglia at the level of the cervical spinal cord of P42 (white arrowheads). h–j, SARS-CoV-2 S (h) and N (i) RNA by ISH and SARS-CoV-2 N (j) protein by chromogenic IHC of cervical spinal cord of P42. k–m, SARS-CoV-2 S (k) and N (l) RNA by ISH and SARS-CoV-2 N (m) protein by chromogenic IHC, all of which are predominantly found in the granular layer (GL) as compared to the molecular level (ML) of cerebellum of P38. WM, white matter. n,o, SARS-CoV-2 S (n) and N (o) RNA by ISH and SARS-CoV-2 N (p) viral protein by chromogenic IHC of basal ganglia of P40. Hoechst 33342 was used to identify nuclei (blue) in all IF images, and IF images were obtained by confocal microscopy. Haematoxylin was used as a counterstain and all ISH and chromogenic IHC images were obtained by bright-field microscopy. Scale bars, 15 μm (a), 10 μm (b–d,g–p) and 25 μm (e,f).
To determine the relationship between SARS-CoV-2 N RNA detected by ddPCR and SARS-CoV-2 spike RNA detected by ISH, we carried out image analysis on interventricular septal tissue from 16 cases covering a four-log range of SARS-CoV-2 N gene copies per nanogram of RNA ddPCR values. Interventricular septum was selected for this analysis owing to consistent histomorphology. Mean SARS-CoV-2 N gene copies per nanogram of RNA significantly correlated with the median SARS-CoV-2 spike RNA-positive cells over thirty ×40 fields (ρ = 0.704, 95% CI: 0.320–0.889, P = 0.002; Supplementary Data 3).
SARS-CoV-2 N RNA and protein in CNS
To further validate detection and distribution of SARS-CoV-2 in CNS tissue, we used a second ISH assay targeting N RNA, and IF and chromogenic IHC-based assays targeting N protein. We confirmed the specificity of these assays with appropriate positive and negative controls (Supplementary Data 3, panels yy–bbb) and applied them to selected CNS tissues that were SARS-CoV-2 positive by ddPCR. We observed SARS-CoV-2 RNA and protein in hypothalamus and cerebellum of an early case (P38) and cervical spinal cord and basal ganglia of late cases (P42 and P40, respectively), with a pattern consistent with neuronal staining (Fig. 3).
COVID-19 histological findings
The histopathology findings from our cohort were similar to those reported in other case series (Extended Data Fig. 5). Of 44 cases, 38 were determined to have died from COVID-19, and of these, 35 (92.1%) had either acute pneumonia or diffuse alveolar damage at the time of death (Supplementary Data 2). Phases of diffuse alveolar damage showed a clear temporal progression (Extended Data Fig. 6). Pulmonary thromboembolic complications were noted in 10 (23%) cases and myocardial infiltrates were observed in four cases, including one case of substantial myocarditis17 (P3). In the lymph nodes and spleen, we observed both lymphodepletion and follicular and paracortical hyperplasia.
Other non-respiratory histological changes were mainly related to complications of therapy or pre-existing comorbidities. Five cases had old ischaemic myocardial scars and three had coronary artery bypass grafts in place. Diabetic nephropathy and steatohepatitis were observed in ten cases (23%) and five cases (12%), respectively. One case had known hepatitis C with cirrhosis, but the other cases of advanced hepatic fibrosis were probably related to fatty liver disease. Hepatic necrosis (13 cases, 30%) and changes consistent with acute kidney injury (17 cases, 39%) were probably related to hypoxic–ischaemic injury in these very ill patients.
In the examination of 11 brains, we found few histopathologic changes, despite substantial viral burden. Vascular congestion was an unusual finding that had an unclear aetiology and could be related to the haemodynamic changes incurred with infection. Global hypoxic–ischaemic change was seen in two cases, one of which was a juvenile (P36) with a seizure disorder who was found to be SARS-CoV-2 positive on hospital admission, but who probably died of seizure complications unrelated to viral infection.
Discussion
Here we provide, to our knowledge, the most comprehensive analysis to date of the cellular tropism, quantification and persistence of SARS-CoV-2 across the human body including the brain. Our focus on short postmortem intervals, a comprehensive standardized approach to tissue collection, dissecting the brain before fixation, preserving tissue in RNAlater and flash freezing of fresh tissue allowed us to detect and quantify SARS-CoV-2 RNA levels with high sensitivity by ddPCR and ISH, as well as isolate virus in cell culture from multiple non-respiratory tissues including the brain, which are notable differences compared to other studies.
We show that SARS-CoV-2 disseminates early in infection in some patients, with a significantly higher viral burden in respiratory than non-respiratory tissues. We demonstrated virus replication in multiple non-respiratory sites during the first two weeks following symptom onset and detected subgenomic RNA in at least one tissue in 14 of 27 cases beyond D14, indicating that viral replication may occur in non-respiratory tissues for several months. Whereas others have proposed that the detection of SARS-CoV-2 in non-respiratory tissues might be due to residual blood within tissues8,18 or cross-contamination from the lungs during tissue procurement8, our data indicate otherwise. Specifically, only 12 of our cases had detectable SARS-CoV-2 RNA in a perimortem plasma sample, only 2 cases had SARS-CoV-2 subgenomic RNA detected in plasma, and negligible, if any, RNA was detected in banked peripheral blood mononuclear cells from representative cases. Further, we validated detection of SARS-CoV-2 outside the respiratory tract by direct cellular identification of virus in cells through ISH, IHC and IF, isolation of SARS-CoV-2 by cell culture, and detection of distinct SARS-CoV-2 spike sequence variants in non-respiratory sites.
Others have previously reported SARS-CoV-2 RNA within the heart, lymph node, small intestine and adrenal gland6,8,9,10,11,12,18. We replicate these findings and conclusively demonstrate that SARS-CoV-2 is capable of infecting and replicating within these and many other tissues, including brain. Specifically, we report the recovery of replication-competent SARS-CoV-2 from thalamus of P38 at D13 using a modified Vero E6 cell line that stably expresses ACE2 and TMPRSS2. This along with detection of genomic RNA and subgenomic RNA through PCR, multiple imaging modalities showing SARS-CoV-2 RNA and protein within cells of the CNS, and distinct minor variants detected through sequencing in the CNS prove definitively that SARS-CoV-2 is capable of infecting and replicating within the human brain.
HT-SGS of SARS-CoV-2 spike demonstrates homogeneous virus populations in many tissues, while also revealing informative virus variants in others. Low intra-individual diversity of SARS-CoV-2 sequences has been observed frequently in previous studies19,20,21, and probably relates to the intrinsic mutation rate of the virus as well as lack of early immune pressure to drive virus evolution. It is important to note that our HT-SGS approach has both a high accuracy and a high sensitivity for minor variants within each sample, making findings of low virus diversity highly reliable22. Genetic compartmentalization of SARS-CoV-2 between respiratory and non-respiratory tissues in several individuals supports independent replication of the virus at these sites, although lack of compartmentalization between sites does not rule out independent virus replication. We note several cases in which brain-derived virus spike sequences showed nonsynonymous changes relative to sequences from other non-CNS tissues. Further studies will be needed to understand whether these cases might represent stochastic seeding of the CNS or differential selective pressure on spike by antiviral antibodies in the CNS, as others have suggested23,24,25.
Our results show that although the highest burden of SARS-CoV-2 is in respiratory tissues, the virus can disseminate throughout the entire body. Whereas others have posited that this viral dissemination occurs through cell trafficking11 due to a reported failure to culture SARS-CoV-2 from blood3,26, our data support an early viraemic phase, which seeds the virus throughout the body following infection of the respiratory tract. Recent work26 in which SARS-CoV-2 virions were pelleted and imaged from plasma of patients with acute COVID-19 supports this mechanism of viral dissemination. Our cohort is predominantly composed of severe and ultimately fatal COVID-19 cases. However, two cases (P36 and P42) reported only mild or no respiratory symptoms and died with, not from, COVID-19, yet had SARS-CoV-2 RNA widely detected across the body and brain. Additionally, P36 was a juvenile with an underlying neurological condition, but without evidence of multisystem inflammatory syndrome in children, suggesting that children may develop systemic infection with SARS-CoV-2 without developing a generalized inflammatory response.
Finally, our work begins to elucidate the duration and locations at which SARS-CoV-2 RNA can persist. Although the respiratory tract was the most common location in which SARS-CoV-2 RNA persisted, ≥50% of late cases also had persistent RNA in the myocardium, lymph nodes from the head and neck and from the thorax, sciatic nerve, ocular tissue, and in all sampled regions of the CNS, except the dura mater. Notably, despite having more than 100 times higher SARS-CoV-2 RNA in respiratory compared to non-respiratory tissues in early cases, this difference greatly diminished in late cases. Less efficient viral clearance in non-respiratory tissues may be related to tissue-specific differences in the ability of SARS-CoV-2 to alter cellular detection of viral mRNA, interfere with interferon signalling, or disrupt viral antigen processing and presentation27,28,29. Understanding mechanisms by which SARS-CoV-2 evades immune detection is essential to guide future therapeutic approaches to facilitate viral clearance.
We detected subgenomic RNA in tissue from more than 60% of the cohort, including in multiple tissues of a case at D99. Although subgenomic RNA is generated during active viral replication, it is less definitive than cell culture at demonstrating replication-competent virus because subgenomic RNA is protected by double-membrane vesicles that contribute to nuclease resistance and longevity beyond immediate viral replication30,31,32,33. However, nonhuman primates exposed to γ-irradiated SARS-CoV-2 inoculum with high subgenomic RNA copy numbers through multiple mucosal routes had detectable SARS-CoV-2 genomic RNA but undetectable subgenomic RNA levels in respiratory samples by day 1 post-inoculation15. These data suggest that detection of SARS-CoV-2 subgenomic RNA probably reflects recent viral replication. Prolonged detection of subgenomic RNA in a subset of our cases may, however, represent defective rather than productive viral replication, which has been described in persistent infection with measles virus—another single-strand enveloped RNA virus—in cases of subacute sclerosing panencephalitis34.
Our study has several important limitations. First, our cohort largely represents older unvaccinated individuals with pre-existing medical conditions who died from severe COVID-19, limiting our ability to extrapolate findings to younger, healthier or vaccinated individuals. Second, our cases occurred during the first year of the pandemic, before widespread circulation of variants of concern, and thus findings might not be generalizable to current and future SARS-CoV-2 variants. Finally, although it is tempting to attribute clinical findings observed in post-acute sequelae of SARS-CoV-2 to viral persistence, our study was not designed to address this question. Despite these limitations, our findings fundamentally improve the understanding of SARS-CoV-2 cellular distribution and persistence in the human body and brain and provide a strong rationale for pursuing future similar studies to define mechanisms of SARS-CoV-2 persistence and contribution to post-acute sequelae of SARS-CoV-2.
Methods
Autopsies
Autopsies were carried out and tissues were collected as previously described35 in the National Cancer Institute’s Laboratory of Pathology at the National Institutes of Health Clinical Center following consent of the legal next of kin. Autopsy patients in this cohort were unvaccinated against SARS-CoV-2. Tissues preserved for histopathologic analysis and special staining were dissected fresh at the time of autopsy, placed into tissue cassettes, fixed for 24 h in neutral-buffered formalin, and then transferred to 70% ethanol for 48 h before impregnation with paraffin.
Measurement of IgG and IgM antibodies to N and spike protein of SARS-CoV-2
Fluid-phase luciferase immunoprecipitation system assays were used to study IgG and IgM antibody response to SARS-CoV-2. For IgG luciferase immunoprecipitation system measurements, Renilla luciferase–nucleocapsid and Gaussia luciferase–spike protein extracts were used with protein A/G beads (Protein A/G UltraLink Resin, Thermo Fisher) as the IgG capture reagent as previously described with microtitre filter plates36. For IgM measurements, anti-human IgM goat agarose beads (Sigma) were substituted as the capture reagent using both the microfilter plate and microtube format37. The IgM immunoprecipitation assays were carried out in 1.5-ml microfuge tube format containing 1 µl serum or plasma, Renilla luciferase–N (10 million light unit input per tube) or Gaussia luciferase–spike protein (40 million light input per tube) and buffer A (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% Triton X-100) to a total volume of 100 µl. After mixing, the tubes were incubated at room temperature for 1 h. Next, 10 µl of the anti-human IgM agarose bead suspension was added to each tube for a further 60 min, and tubes were placed on a rotating wheel at 4 °C. The samples were then washed by brief centrifugation to collect the bead pellet at room temperature three times with 1.5 ml buffer A and once with 1.5 ml PBS. After the final wash, the beads were mixed with coelenterazine substrate (100 µl) and light units were measured in a tube luminometer. Known seronegative and seropositive samples for IgG and IgM antibodies to the N and spike proteins were used for assigning seropositive cutoff values and for standardization.
SARS-CoV-2 RNA quantification of tissues and body fluids
Total RNA was extracted from RNAlater (Invitrogen)-preserved tissues and body fluids collected at autopsy using the RNeasy Mini, RNeasy Fibrous Tissue Mini, RNeasy Lipid Tissue Mini and QIAamp Viral RNA Mini Kits (Qiagen) according to the manufacturer’s protocols. Upstream tissue processing and subsequent RNA quantification have been described previously35. The QX200 AutoDG Droplet Digital PCR System (Bio-Rad) was used to detect and quantify SARS-CoV-2 RNA in technical replicates of 5.5 µl RNA for fluids and up to 550 ng RNA for tissues. Raw data were collected using QuantaSoft version 1.7.4.0917 and analysed using QuantaSoft Analysis Pro version 1.0.596. Results were then normalized to copies of N1, N2 and RP per millilitre of sample input for fluids and per nanogram of RNA concentration input for tissues. Samples had to be positive for the human RNase P (RP) gene at the manufacturer’s limit of detection (LOD) of ≥0.2 copies per microlitre and ≥4 positive droplets per well to ensure RNA extraction was successful and be reported. For samples to be considered positive for SARS-CoV-2 N1 or N2 genes, the technical replicates needed to have an average at or above the manufacturer’s LOD of ≥0.1 copies per microlitre and ≥2 positive droplets per well. More than 60 control autopsy tissues from uninfected individuals, representing all organs collected for COVID-19 autopsy cases, were used to validate the manufacturer’s emergency use authorization published LOD for nasopharyngeal swabs for tissues (Supplementary Data 1e). ddPCR data for P3 (ref. 17) as well as a portion of the oral cavity35 have been reported previously.
Subgenomic RNA analysis of ddPCR positive tissues
Tissues that tested positive for one or both SARS-CoV-2 N gene targets through ddPCR had RNA submitted for subgenomic RNA analysis. Briefly, 5 µl of sample RNA was added to a one-step real-time RT–qPCR assay targeting subgenomic RNA of the envelope (E) gene (forward primer 5′-CGATCTCTTGTAGATCTGTTCTC-3′; reverse primer 5′-ATATTGCAGCAGTACGCACACA-3′; probe 5′-FAM-ACACTAGCCATCCTTACTGCGCTTCG-ZEN-IBHQ-3′)38 using the Rotor-Gene probe kit (Qiagen) according to instructions of the manufacturer. In each run, standard dilutions of counted RNA standards were run in parallel to calculate copy numbers in the samples. The LOD for this assay was determined to be <40 Cq (Supplementary Data 1) using 40 control autopsy tissues from uninfected individuals, representing all organs collected for COVID-19 autopsy cases.
Virus isolation from select postmortem tissues
Select tissues with high viral RNA levels through ddPCR and subgenomic RNA RT-qPCR measuring across a broad range of 16 to <35 Cq underwent virus isolation to prove the presence of infectious virus. Virus isolation was carried out on tissues by homogenizing the tissue in 1 ml DMEM and inoculating Vero E6 cells in a 24-well plate with 250 µl of cleared homogenate and a 1:10 dilution thereof. Plates were centrifuged for 30 min at 1,000 r.p.m. and incubated for 30 min at 37 °C and 5% CO2. The inoculum was then removed and replaced with 500 µl DMEM containing 2% FBS, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin. Six days after inoculation, the cytopathic effect was scored. A blind passage of samples in which no cytopathic effect was present was carried out according to the same method. Additional virus isolation from P38 thalamus and hypothalamus was carried out using Vero E6-TMPRSS2-T2A-ACE2 (catalogue no. NR-54970, BEI Resources) grown in DMEM containing 10% FBS, 50 U ml−1 penicillin, 50 μg ml−1 streptomycin and 10 μg ml−1 puromycin. Virus isolation was carried out as described for other tissues on Vero E6 cells, without selection antibiotics. Tissue homogenate from flash-frozen specimens and supernatants from plates were analysed using RT–qPCR for SARS-CoV-2 E gene subgenomic RNA (described above) or genomic RNA as previously described39 to rule out other causes for the cytopathic effect. Cell lines were not authenticated in house, but were confirmed to be free of mycoplasma contamination.
Virus sequencing
Five early cases (P18, P19, P27 and P38) and one late case (P33) with multiple body site tissues containing subgenomic RNA levels ≤31 Cq were selected for HT-SGS as previously described22. Presence of variants of SARS-CoV-2 was analysed within and between tissues.
Supernatant from the virus isolation plates of thalamus of P38 were sequenced using short-read, whole-genome sequencing. Total RNA was depleted of rRNA using Ribo-Zero+ following the manufacturer’s protocol (Illumina). Cleaned RNA was eluted in water and sequencing libraries were prepared following the Kapa RNA HyperPrep kit according to the manufacturer’s protocol (Roche Sequencing Solutions). Briefly, 10 µl of depleted RNA was used as a template for fragmentation (65 °C for 1 min) and first-strand synthesis. To facilitate multiplexing, adapter ligation was carried out with KAPA Unique Dual-Indexed Adapters, and samples were enriched for adapter-ligated product using KAPA HiFi HotStart Ready mix and a range of 9–19 PCR amplification cycles based on SARS-CoV-2 Ct values and total RNA starting inputs. Pools consisting of 1–6 sample libraries were used for hybrid-capture virus enrichment using myBaits Expert Virus SARS-CoV-2 panel following the manufacturer’s manual, version 5.01, with a range of 12–18 cycles of post-capture PCR amplification (Arbor Biosciences). Purified, enriched libraries were quantified on a CFX96 Real-Time System (Bio-Rad) using Kapa Library Quantification kit (Roche Sequencing Solutions). Libraries were diluted to 2 nM stock, pooled together as needed in equimolar concentrations and sequenced on the MiSeq (Illumina) generating 2 × 150-bp paired-end reads. Raw sequence reads were trimmed of Illumina adapter sequence using Cutadapt version 1.12 (ref. 40) and then trimmed and filtered for quality using the fastq_quality_trimmer and fastq_quality_filter tools from the FASTX-Toolkit 0.0.14 (Hannon Lab, CSHL). Reads were then mapped to the SARS-CoV-2 2019-nCoV/USA-WA1/2020 genome (MN985325.1) using Bowtie2 version 2.2.9 (ref. 41) with parameters -local -no-mixed -X 1500. PCR duplicates were removed using picard MarkDuplicates, version 2.26.10 (Broad Institute).
SARS-CoV-2 RNA ISH
Chromogenic ISH detection was carried out using the manual RNAScope 2.5 HD assay (catalogue no. 322310, Advanced Cell Diagnostics) with a modified pretreatment protocol. Briefly, formalin-fixed and paraffin-embedded (FFPE) tissue sections were cut at 7 μm, air dried overnight, and baked for 1–2 h at 60 °C. The FFPE tissue sections were deparaffinized, dehydrated and then treated with pretreat 1 for 15 min at room temperature. The slides were boiled with pretreatment reagent for 15 min, digested with protease at 40 °C for 20 min, and then hybridized for 2 h at 40 °C with probe-V-nCov2019-S (catalogue no. 848561, Advanced Cell Diagnostics) or probe-V-nCoV-N (catalogue no. 846081, Advanced Cell Diagnostics)42. In addition, probe-Hs-PPIB (peptidylprolyl isomerase B, catalogue no. 313901, Advanced Cell Diagnostics) and probe-dapB (catalogue no. 310043, Advanced Cell Diagnostics) were used as a positive and negative control, respectively. Subsequent amplification was carried out according to the original protocol. Detection of specific probe-binding sites was visualized with RNAScope 2.5 HD Reagent Kit-BROWN chromogenic labels (Advanced Cell Diagnostics). The slides were counterstained with haematoxylin and coverslipped.
To correlate viral load detected between ddPCR and ISH, the interventricular septum of 16 cases spanning a four-log range of SARS-CoV-2 N copies per nanogram of RNA through ddPCR underwent ISH and subsequent quantification using image analysis. The interventricular septum stained slides were digitalized using a NanoZoomer XR Digital Pathology system (Hamamatsu, Hamamatsu City, Japan) at 40× magnification. Digitalized images were automatically analysed using Visiopharm software v2021.09.02 (Visiopharm, Hørsholm, Denmark). A training set was used to configure the algorithm and identify SARS-CoV-2 RNA signals. In brief, 3,3′-diaminobenzidine (DAB) dots of positive signals were identified using a Bayesian classifier trained on pre-processing steps. We randomly selected 30 regions of interest per slide and calculated the median of positive cells.
SARS-CoV-2 multiplex IF
FFPE CNS sections were deparaffinized, rehydrated and subjected to 0.01 M citrate buffer antigen retrieval for 20 min at 120 °C. Slides were then rinsed briefly in deionized water, washed with PBS and subsequently incubated in a 5% milk (catalogue no. 1706404, Bio-Rad), 5% normal donkey serum, 0.3 M glycine and 0.1% Triton X-100 blocking solution made up in PBS for 30 min. Primary antibodies to SARS-CoV-2 N protein 1 (NP1, 1:1,000, custom made GenScript U864YFA140-4/CB2093)43,44,45 and neuronal nuclear protein (NeuN, 1:200, catalogue no. MAB377, Chemicon) or transmembrane protein 119 (TMEM119, 1:1,000, catalogue no. MAB130313, R&D Systems) were diluted in blocking serum and applied to slides overnight at 4 °C. The following day, slides were washed extensively with PBS to remove any detergent and freshly made True Black Plus solution (1:40 in PBS, catalogue no. 23014, Biotium) was applied for 14 min. Slides were again extensively washed and then species-specific secondary conjugates (1:500, catalogue nos. A-21206 and A-21203, Thermo Fisher) were applied for 1 h at room temperature. Following PBS wash, Hoechst 33342 was applied for 10 min (1:2,000, catalogue no. H3570, Thermo Fisher) to label nuclei. Slides were coverslipped with Prolong Gold (catalogue no. P36930, Thermo Fisher).
SARS-CoV-2 chromogenic IHC
Chromogenic IHC was carried out on various ddPCR positive CNS and lung tissues and negative pre-pandemic control cases demonstrating relative expression of the target protein between infected and control samples. Briefly, 5 µm FFPE tissue sections were incubated at 60 °C for 2 h, deparaffinized in xylene, and hydrated in serial alcohol solutions to distilled water. Heat antigen retrieval was carried out using a pressure cooker (DAKO) by submerging slides in 1× pH 6 citrate buffer for 20 min. Endogenous enzyme activity was quenched with 3% hydrogen peroxide containing sodium azide for 10 min with additional 10% non-fat dry milk (Bio-Rad) for 20 min to prevent nonspecific binding. Tissue sections were then incubated with polyclonal SARS or SARS-CoV-2 N antibody (1:500, custom made, GenScript U864YFA140-4/CB2093, 0.447 mg ml−1)43,44,45 for 1 h at room temperature. Negative controls were congruently stained on subsequent sections following the same protocol replacing the primary antibody with a rabbit IgG control antibody (0.5 mg ml−1, catalogue no. I-1000-5, Vector Laboratories). The antigen–antibody reaction was detected with Dako Envision+Rb polymer detection system (DAKO) and visualized with DAB chromogen. Sections were lightly counterstained with haematoxylin, dehydrated in graded alcohols, cleared in xylene, mounted and coverslipped.
Statistical analysis
Correlations between two continuous variables were assessed using Spearman’s rank correlation coefficient (ρ). Fisher’s z-transformation was used for the calculations of 95% CI and P values. To compare ddPCR levels between tissue types (respiratory versus non-respiratory), we used linear mixed models with compound symmetry correlation structure to account for repeated measures within each subject. Standard residual diagnoses were used to check model assumptions. Log-transformations were used when needed. To log10-transform ddPCR values, 0 values were replaced with a small positive random number according to the detection limits. Logistic regression models were used to generate ROC curves. Optimal cutoff values were selected by treating sensitivity and specificity as equally important. SAS version 9.4 was used for all analyses. All P values are two-sided and reported without adjustment for multiple comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Quelle: nature
Den ersten Kommentar schreiben.
Antworten
Sie müssen eingeloggt sein, um einen Kommentar zu schreiben.